
Depending on the library preparation method, the RNA Seq data can be stranded or non-stranded. Here, is the link from Azenta, to better understand stranded vs non-stranded library preparation. Here is the screenshot taken from Hong Zheng which describes about the strandedness.
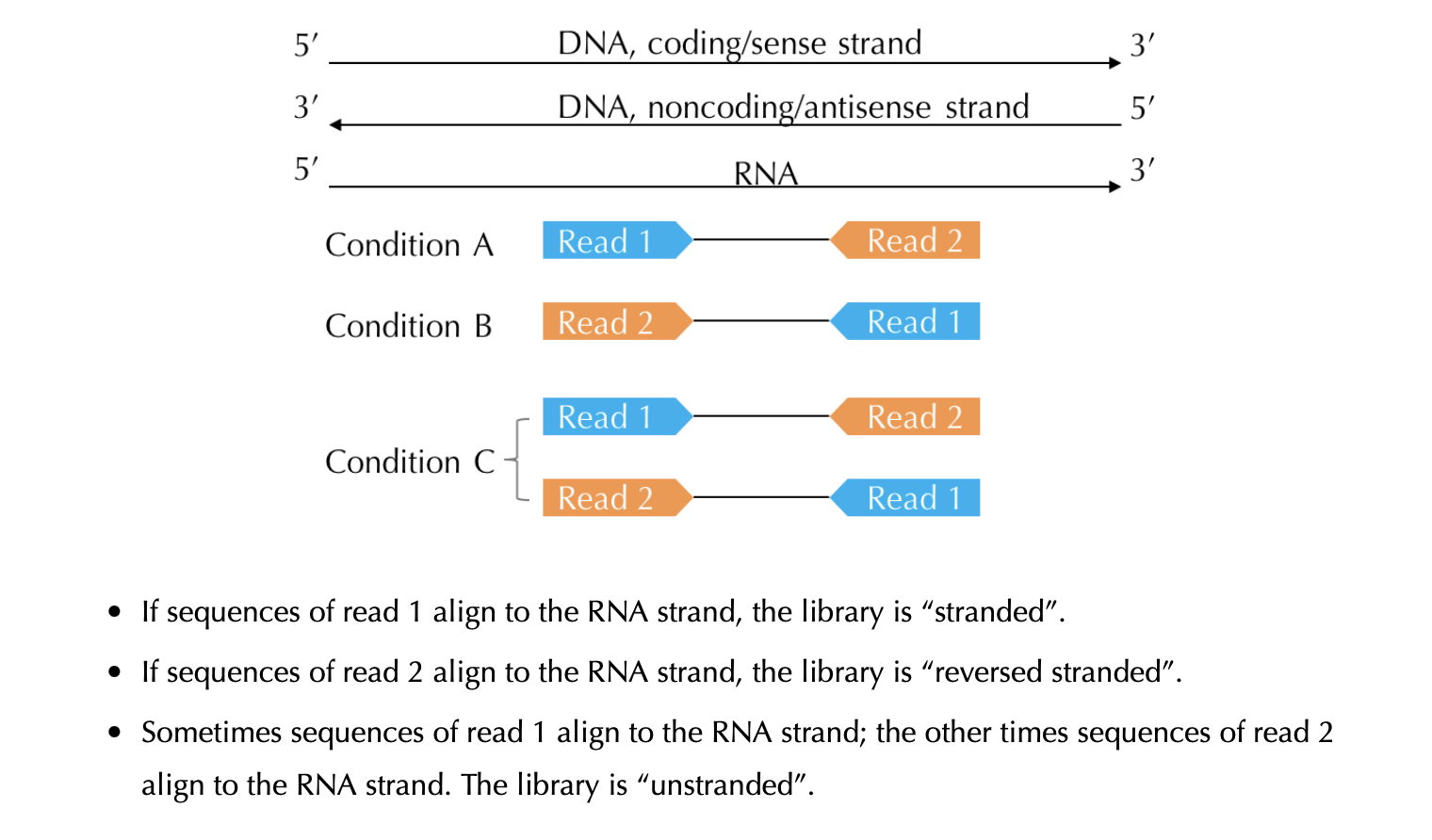
Since different tools uses strand information to align and get the read counts for the genes, it is important to know the strandedness of RNA seq data. Mostly, for the public data strand information is not provided. The strandedness can be determined using the tool RSeQC. infer_experiment.py can be used to identify the strandedness. For details visit this link. Here, is the command example:
1
2
3
4
5
6
7
8
infer_experiment.py -r hg19.refseq.bed12 -i Pairend_nonStrandSpecific_36mer_Human_hg19.bam
#Output::
This is PairEnd Data
Fraction of reads failed to determine: 0.0172
Fraction of reads explained by "1++,1--,2+-,2-+": 0.4903
Fraction of reads explained by "1+-,1-+,2++,2--": 0.4925
If both 1++,1--,2+-,2-+ and 1+-,1-+,2++,2-- has equal proportion. The RNA Seq data is most likely unstranded. If 1++,1--,2+-,2-+ has more proportion, it is most likely forward stranded (FR, F1R2) else reverse stranded (RF, F2R1).
There is another tool called how_are_we_stranded_here, which can also be used to identify the strandedness. On the background it also uses RSeQC. For RSeQC, you will need bam file, but for how_are_we_stranded_here you can directly use fastq files.
The details regarding diffrent tools and their parameters for strandedness is provided here in this link from griffith lab.