
UCSC table browser is a great tool that can be used to download different annotation data such as exon, intron, 5’ UTR, 3’ UTR etc.
Here is an example to extract the exons from human genome version hg19:
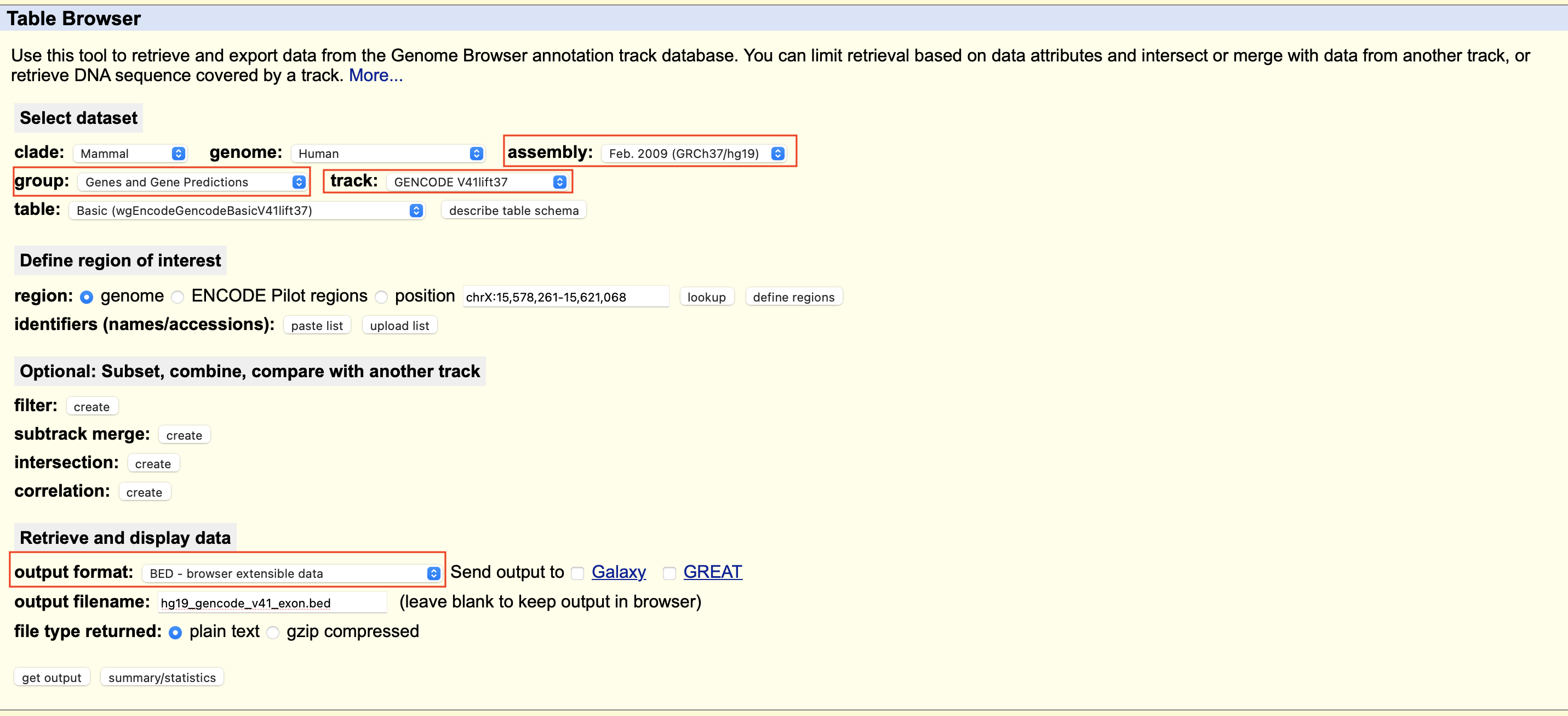
- Select genome to
Human - Select group as
Genes and Predictions - Select your desired track. I have chosen
GENCODE V41lift37. - Select region to
positionif you have specific region of interest else, keep it atgenome. - Select output format as
BED. This will generate output in bed format with each row representing as one exonic region. - Give your output file name
- Select the output file type and then click
get output. This will open another window, where you can select which genome region you want to extract. Look at the figures below for details.
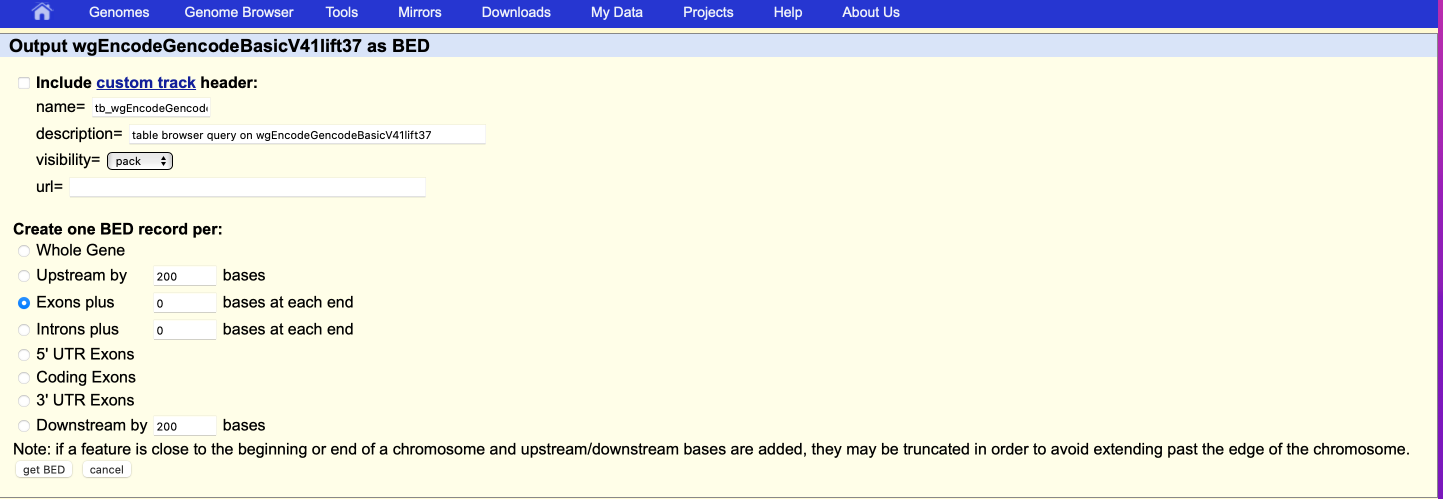
You can also download the gtf file, and parse the file to get the desired information such as TSS, intergenic region, exons, introns etc. You need to install bedtools for this.
Extracting intergenic region
1
awk 'BEGIN{OFS="\t";} $3=="gene" {print $1,$4-1,$5}' hg19_gencode.v41lift37.annotation.gtf | sortBed | complementBed -i stdin -g hg19_chrom.sizes > gencode.v41.intergenic_region.bed
Extracting exon coordinates
1
awk '{OFS="\t"} $3=="exon" {print $1,$4-1,$5}' hg19_gencode.v41lift37.annotation.gtf | sortBed | bedtools merge -i stdin > hg19_gencode_v41_exon.bed
Extracting intron coordinates
1
awk '{OFS="\t"} $3=="gene" {print $1,$4-1,$5}' hg19_gencode.v41lift37.annotation.gtf | sortBed | bedtools subtract -a stdin -b stdin_test.bed > hg19_gencode_v41_intron.bed
The hg38_chrom.sizes can be downloaded from this link. Or you can generate yourself from the fasta file.
1
2
3
4
5
6
7
# First create index file using samtools
samtools faidx hg19.fa
# Extract column 1 and 2 from genome index file
cut -f1,2 hg19.fa.fai > hg19_chrom.sizes