
There are several tools available map sequencing data to the reference genome. And for all of the tools index files needs to be generated first. Here are some of the tools and the syntax used to generate index file.
DNA Sequence
Burrows-Wheeler Alignment Tool (bwa)
1
2
3
bwa index hg38.fa
samtools faidx hg38.fa
cut -f1,2 hg38.fa.fai > hg38.sizes
1
bowtie2-build hg38.fa index_filename
RNA Sequence
Spliced Transcripts Alignment to a Reference (STAR)
1
STAR --runThreadN 20 --runMode genomeGenerate --genomeDir index_files/star_2.5.3a --genomeFastaFiles hg38.fa --sjdbGTFfile gencode.v41.annotation.gtf --sjdbOverhang 99
sjdbOverhang value = readlength - 1
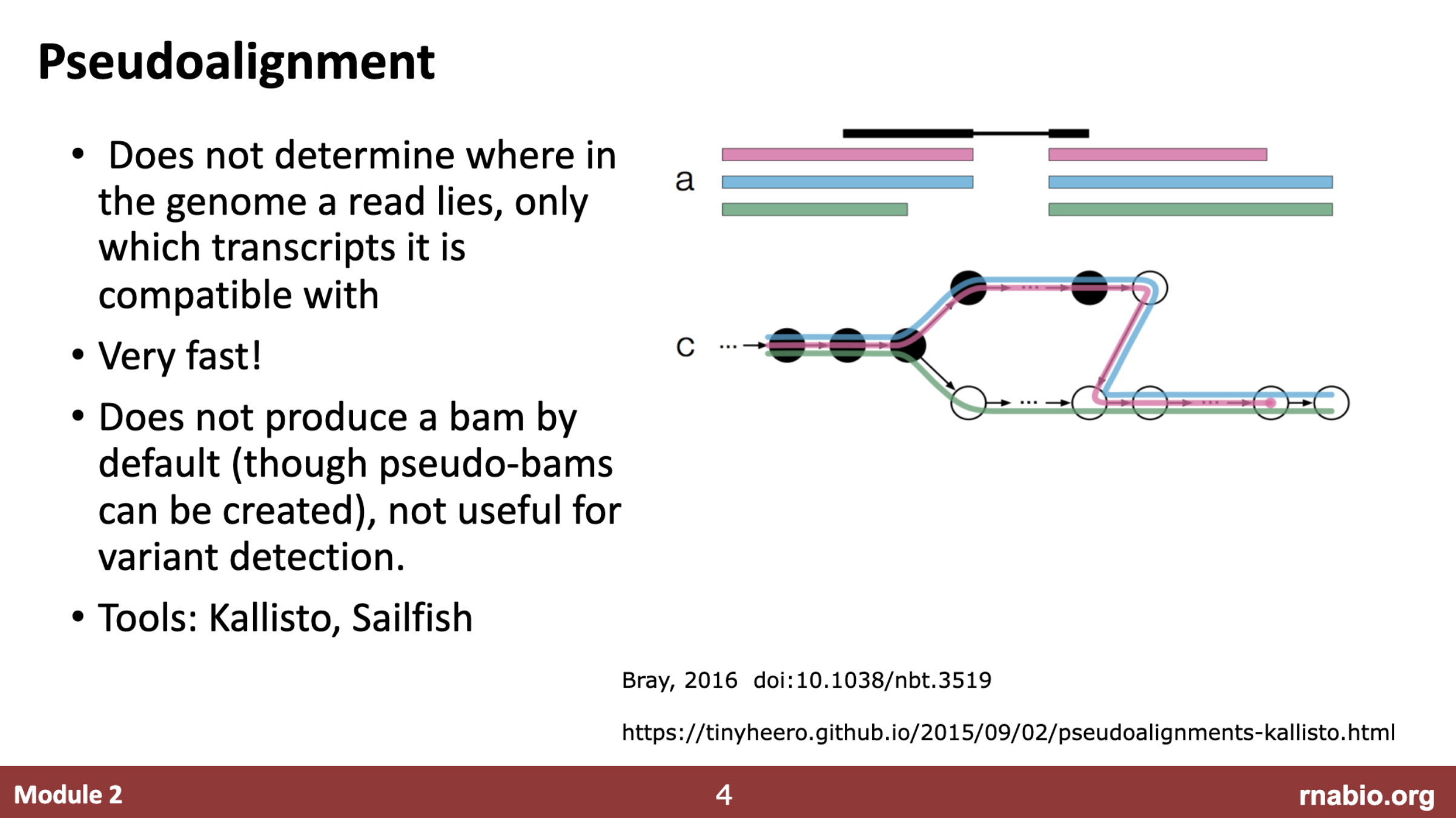
kallisto doesn’t allign sequencing reads to the genome, it performs pseudoalignment for transcript quantification using indexed transciptome. You can download the cdna fasta sequence from ensembl site.
1
kallisto index -i index_filename hg38.cdna.fa
example: index_filename = hg38_index.idx
Reference
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4631051/
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3322381/