
To find the closest gene to the given genomic region, we will be using bedtools closest. Fist lets see our region of interest
1
2
3
4
5
6
7
8
9
10
11
12
13
14
$ cat input_region.bed
chr16 566689 566964 id . +
$ head -n 8 gencode.hg38.v41.bed
chr1 14404 29570 ENSG00000227232.5 WASH7P -
chr1 17369 17436 ENSG00000278267.1 MIR6859-1 -
chr1 29554 31109 ENSG00000243485.5 MIR1302-2HG +
chr1 30366 30503 ENSG00000284332.1 MIR1302-2 +
chr1 34554 36081 ENSG00000237613.2 FAM138A -
chr1 52473 53312 ENSG00000268020.3 OR4G4P +
chr1 57598 64116 ENSG00000240361.2 OR4G11P +
chr1 65419 71585 ENSG00000186092.7 OR4F5 +
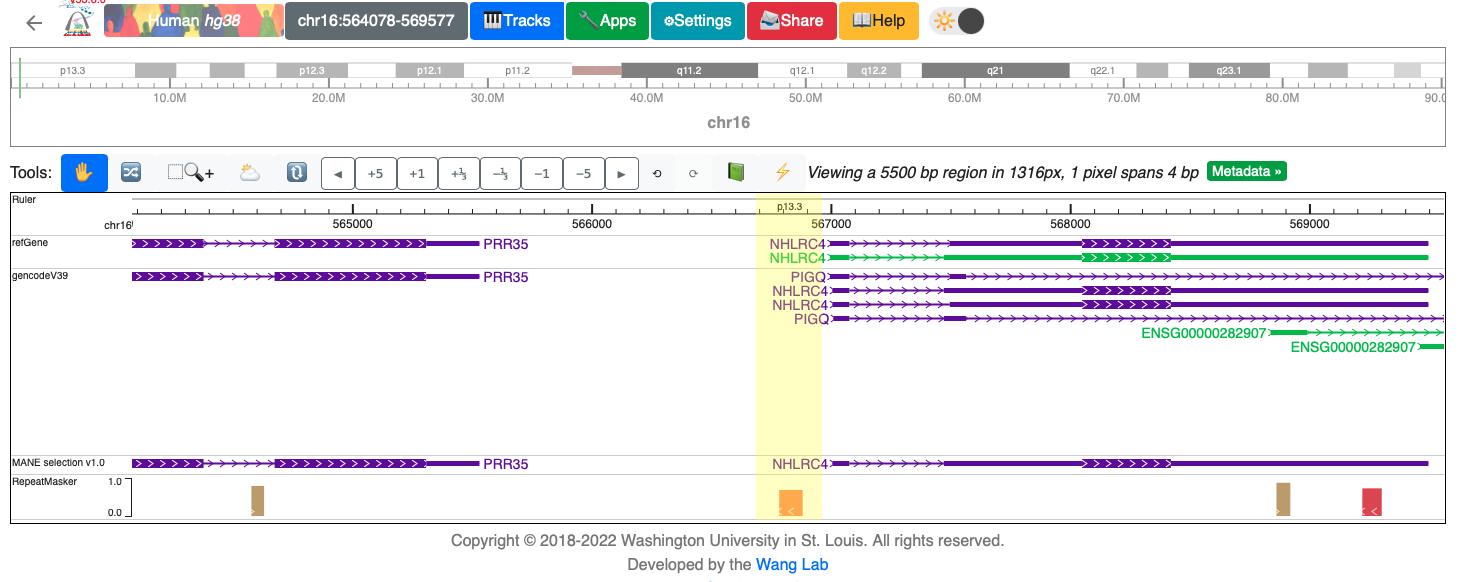
Our region of interest is highlighted in yellow in the figure below, and there are 3 nearby genes (NHLRC4, PIGQ and PRR35). We can run the bedtools closest to get the nearest gene.
1
2
3
$ bedtools closest -a input_region.bed -b gencode.hg38.v41.bed -D ref -t all
chr16 566689 566964 id . + chr16 566995 584109 ENSG00000007541.17 PIGQ + 32
Here, using -D ref assigns input file (input_region.bed) used in -a parameter as reference. -t all used to report all the genes incase two genes are at exact same distance. But, we can see that although both NHLRC4 and PIGQ are very close to our region, only the closest is reported. We can use parameter -k to report to increase the number of nearest genes to be reported.
1
2
3
4
5
6
$ bedtools closest -a input_region.bed -b gencode.hg38.v41.bed -D ref -t all -k 3
chr16 566689 566964 id . + chr16 566995 584109 ENSG00000007541.17 PIGQ + 32
chr16 566689 566964 id . + chr16 567005 569495 ENSG00000257108.2 NHLRC4 + 42
chr16 566689 566964 id . + chr16 560394 565529 ENSG00000161992.6 PRR35 + -1161
The last column represents the distance from our region of interest. Negative value means upstream and positive means downstream. To get more number of nearest genes, you can just increase the value in the -k, so that you don’t miss any genes if there are huge gene clusters in your region of intereset. But it also causes the addition of genes which are very far away. In this case, you can just filter the genes based on your desired distance.
Lets say you want the genes which are less than 42 bp in downstream and less than 1200 bp in upstream. You can do it by simply using awk.
1
2
3
4
$ bedtools closest -a input_region.bed -b gencode.hg38.v41.bed -D ref -t all -k 3 | awk '($13 < 42 && $13 > -1200)'
chr16 566689 566964 id . + chr16 566995 584109 ENSG00000007541.17 PIGQ + 32
chr16 566689 566964 id . + chr16 560394 565529 ENSG00000161992.6 PRR35 + -1161